
Design of DNA sequences for DNA computing
Coordinator/contact:
This email address is being protected from spambots. You need JavaScript enabled to view it.
This email address is being protected from spambots. You need JavaScript enabled to view it.
Introduction
The design of reliable DNA sequences is crucial in many engineering applications which depend on DNA-based technologies, such as nanotechnology or DNA computing. In these cases, two of the most important properties that must be controlled to obtain reliable sequences are self-assembly and self-complementary hybridization. These processes have to be restricted to avoid undesirable reactions, because undesirable reactions usually lead to incorrect computations. Therefore, it is important to design robust sets of sequences which only interact with the sequences they are supposed to interact when specific bio-chemical conditions are reached. The design of reliable DNA sequences involves several heterogeneous and conflicting design criteria that do not fit traditional optimization methods, therefore DNA sequence design can be naturally modelled as a multiobjective optimization problem. As a general rule, the more biochemical criteria considered, the more reliable are the sequences generated. However, it is not computationally practical to include a very high number of design criteria, and some of them even overlap with others, so it is necessary to study carefully which objectives will be used to model the problem. After revising related publications, we have chosen six biochemical criteria. Four of them are taken as objectives that have to be simultaneously optimized by using a multiobjective evolutionary algorithm (MOEA), and the other two are taken as constraints that every generated sequence has to satisfy. The objectives are: continuity, hairpin, similarity and h-measure (all of them to be minimized), and the constratins are GC ratio and melting temperature.
DNA sequence design problem
DNA sequence design problem consists of designing sets of reliable sequences (named DNA libraries) which form stable duplexes while avoiding interactions of undesirable sequences. Every criterion used to design sequences should contribute to improve reliability, because this property is a mandatory requirement for any system based on DNA sequences. There are several biological criteria that can be considered to achieve this purpose. According to their biological meaning, design criteria can be classified into four groups. First, properties that avoid inconvenient reactions (h-measure and similarity design criteria are included in this category). Second, criteria that control the generation of secondary structures (continuity and hairpin belong to this group). Third, properties that control the biochemical characteristics of DNA sequences (GC content and melting temperature design criteria are two of the most representative properties in this category). And finally, criteria that restrict the sequences composition (e.g.: sequences with four ‘A’ bases in a row are not allowed, or it is mandatory that all generated sequences include a specific subsequence with the purpose of controlling a specific biological reaction).
H-measure
This objective is very important, because it prevents cross hybridization between sequences. H-measure is calculated by computing how many nucleotides are complementary between the sequences in a generated set. Sequences in this set are supposed to not interact with each other, so H-measure is an objective to be minimized. To provide a more reliable measure, elongated sequences with gaps are considered. The mathematical definition for this objective is described as follows:
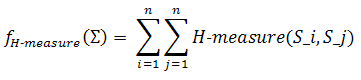
where S_i and S_j are anti-parallel sequences and n is the number of sequences which are in the set of DNA sequences which is being considered. Moreover, H-measure is divided into two terms. One is the penalty for the continuous complementary region and the other for the overall complementarily. Formally, this measure is expressed as follows:
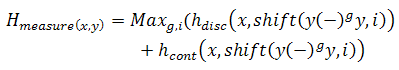
where shift indicates a shift of i bases in the sequence y. Moreover, the sequence y is duplicated and between the two copies of the sequence are located some gaps, symbolized by (–). The number of gap spaces between sequences is between 0 and l – 3, 0 <= g <= l – 3, where l is the length of sequences, and |i| = l. Therefore, the expression y(–)gy indicates g gaps between the y sequence and its copy.
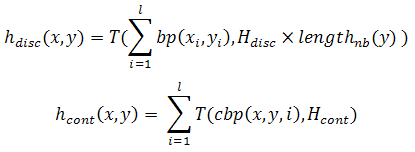
In the previous equation, Hdisc is a real value between 0 and 1; Hcont is an integer between 1 and l; cbp(x,y,i) measures the length of continuous basepairing from the ith base of sequence x; bp(xi, yi) is equal to 1 if xi = !yi and equal to 0 otherwise. Finally, T(i, j) = i if i > j and 0 otherwise.
Similarity
This objective computes, including shift positions, the similarity in the same direction of two given sequences with the aim of keeping each sequence as unique as possible. A more detailed comparison is performed by extending the target sequence with its own sequence to the 3’-end. Moreover, variable position gaps are also included between the sequence and its copy. Continuous and discontinuous similarities are also considered. In the first case, the same substring appears in two sequences, while in the discontinuous case, the overall trend of two sequences resembles each other. For example, the sequences 5’-GCATATGCCGAT-3’ and 5’-GGCCCTGCCAAA-3’ have a continuous similarity of 4 (the substring TGCC, from the 6th to the 9th base), and a discontinuous similarity (without considering position shifts) of 6. The mathematical definition for this measure is described in the following equation:
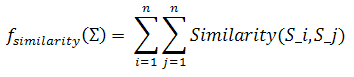
where S_i and S_j are parallel DNA sequences and n is the number of sequences which are in the set of DNA sequences which is being considered. Moreover, Similarity(S_i, S_j) is also divided into two terms. One is for the overall discrete discontinuous similarity of every pair of sequences (x,y) within the DNA library, and the other is the penalty for the continuous common subsequence.
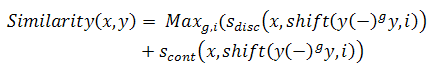
where shift indicates again a shift of sequence y by i bases, (–) denotes a gap, 0 <= g <= l – 3, where l is the length of sequences, and |i| = l.
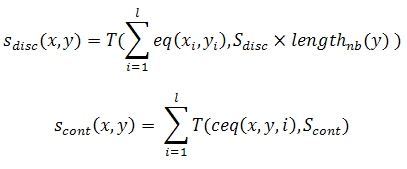
where Sdisc is a real value between 0 and 1; Scont is an interger between 1 and l; ceq(x,y,i) measures the length of continuous common subsequence starting from the ith base of sequence x; eq(xi, yi) is equal to 1 if xi = yi and 0 otherwise. Finally, T(i, j) = i if i > j and 0 otherwise.
Continuity
This measure indicates the degree of successive occurrences of the same base in a sequence. The measure prohibits consecutive runs of the same nucleotide over a given threshold. For example, if the threshold is 4, in the sequence TACGGTAACTAAAAACGTCGAAATGCGGGCACT, the third subsequence of adenines (A) violates the continuity and the others not. The mathematical definition for this measure is expressed in the following equation:
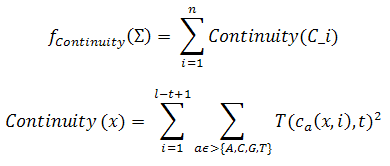
where n is the number of sequences which are in the set of DNA sequences which is being considered and Ca(x,i) is equal to epsilon if there is an epsilon, s.t. xi is different from a, xi+j = a, for 1 <= j <= epsilon, xi+epsilon+1 is different from a, and 0 otherwise.
Hairpin
This criterion indicates the probability that the DNA sequence under study can create secondary structures. This objective is calculated through the Hamming distance by considering the length of hairpin loop and the number of hybridized pairs. It is assumed that a hairpin has at least Rmin bases as a loop and a minimum of Pmin base pairs as a stem. The penalty for different-size hairpin formation at every position in the sequence under study is also considered. In the following equation, we consider hairpins with r-base loop and p-base pairs stem to be formed at position i in the sequence x, if more than half bases in the subsequence xi-p...xi hybridize to the subsequence xi+r...xi+r+p. The number of matches in these subsequences is defined as the penalty for this hairpin.
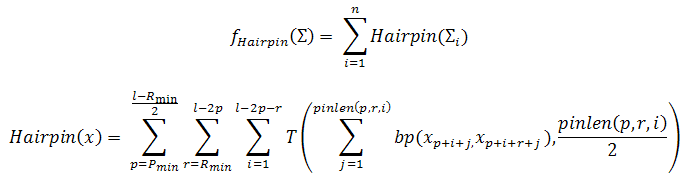
where n is the number of sequences which are in the set of DNA sequences which is being considered and the function pinlen (p,r,i) = min(p+i, l–r–i–p) and denotes the maximum number of possible basepairs when a hairpin is formed with centre in p+i+r/2.
GC content
This criterion indicates the percentage of bases C and G in the sequence x. This is important because the GC content affects to the chemical properties of DNA sequences. GC base pairs are more stable than AT base pairs because the GC pair is bound by three hydrogen bonds, while AT pairs are bound by two hydrogen bonds. This fact makes high-GC-content DNA structures more tolerant of high temperatures. For example, if we consider the sequence ATGATAGGCGTTGTA, the GC% is 40 (6 out of 15).
Melting temperature
This criterion predicts DNA thermal denaturation, which is a key factor for DNA computing. It is desirable that DNA sequences have similar chemical features for successful operations. Both sequence and base composition are important determinants of DNA duplex stability. DNA melting is the process by which double-stranded DNA unwinds and separates into single-stranded strands (breaking of hydrogen bonding between the bases). There are many ways to calculate this relevant feature, but we use the nearest neighbour (NN) model. The mathematical description for this measure, calculated by using the NN method, is expressed as follows:
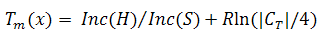
where x is the DNA sequence studied, R is a gas constant and |CT| is the total sequence concentration. Inc(H) and Inc(S) refer to predicted enthalpies and entropies.
Multiobjective formulation
DNA sequence design problem can be naturally formulated as a multiobjective optimization problem and solved by using a multiobjective evolutionary algorithm (MOEA). The problem has been formulated by considering four objectives (similarity, h-measure, continuity and hairpin) and two constraints (melting temperature and GC content), and it is expressed as follows.
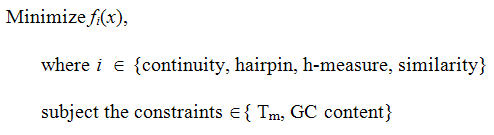
Instances
We use three sets of DNA sequences with different sizes. These datasets were proposed by different authors and they have been successfully applied to reliable DNA computing in several studies. These instances with different number of sequences and different sizes (nucleotides per sequence) ensure that our algorithms are able to work with different types of instances that have been tested for bio-molecular computing. In the following figure is illustrated each data set used.

References
"DNA Strand Generation for DNA Computing by Using a Multi-Objective Differential Evolution Algorithm". J.M. Chaves-González, M.A. Vega-Rodríguez. BioSystems, Volume 116, Elsevier, 2014, pp. 49–64. ISSN: 0303-2647. (Impact Factor = 1.548)
"A Multiobjective Approach Based on the Behaviour of Fireflies to Generate Reliable DNA Sequences for Molecular Computing". J.M. Chaves-González, M.A. Vega-Rodríguez. Applied Mathematics and Computation, Volume 227, Elsevier, 2014, pp. 291–308. ISSN: 0096-3003. (Impact Factor = 1.551)
"A Multiobjective Swarm Intelligence Approach based on Artificial Bee Colony for Reliable DNA Sequence Design". J.M. Chaves-González, M.A. Vega-Rodríguez, J.M. Granado-Criado. Engineering Applications of Artificial Intelligence, Volume 26, Issue 9, Elsevier, 2013, pp. 2045–2057. ISSN: 0952-1976. (Impact Factor = 1.962)
"DNA Base-code Generation for Reliable Computing by Using Standard Multi-objective Evolutionary Algorithms". J.M. Chaves-González, M.A. Vega-Rodríguez. In: Proceedings of the Genetic and Evolutionary Computation Conference Companion (GECCO 2013), ACM, 2013, pp. 1617–1624.
"DNA Sequence Design for Reliable DNA Computing by Using a Multiobjective Approach". J.M. Chaves-González, M.A. Vega-Rodríguez. In: Proceedings of the 13th IEEE International Symposium on Computational Intelligence and Informatics (CINTI 2012), IEEE, 2012, pp. 73–78.